
Posted by SMCS-Psi Data Analytics Pvt. Ltd. | Pune & Leipzig
In the rapidly evolving world of pharmaceuticals, traditional drug discovery methods are often time-consuming, costly, and limited in scope. At SMCS-Psi Data Analytics Pvt. Ltd., we are redefining this process through the power of machine learning (ML) and artificial intelligence (AI).
Our latest initiative brings together computational biology, cheminformatics, and deep learning to transform how new therapeutic compounds are identified, evaluated, and optimized.
The Vision: Smarter, Faster Drug Discovery
Drug discovery is a complex and multi-stage journey—from identifying biological targets and screening compounds to assessing binding affinities, and finally to testing efficacy and safety. Our team at SMCS-Psi is harnessing ML to automate and enhance these critical stages, reducing the development timeline and increasing the success rate of potential drug candidates.
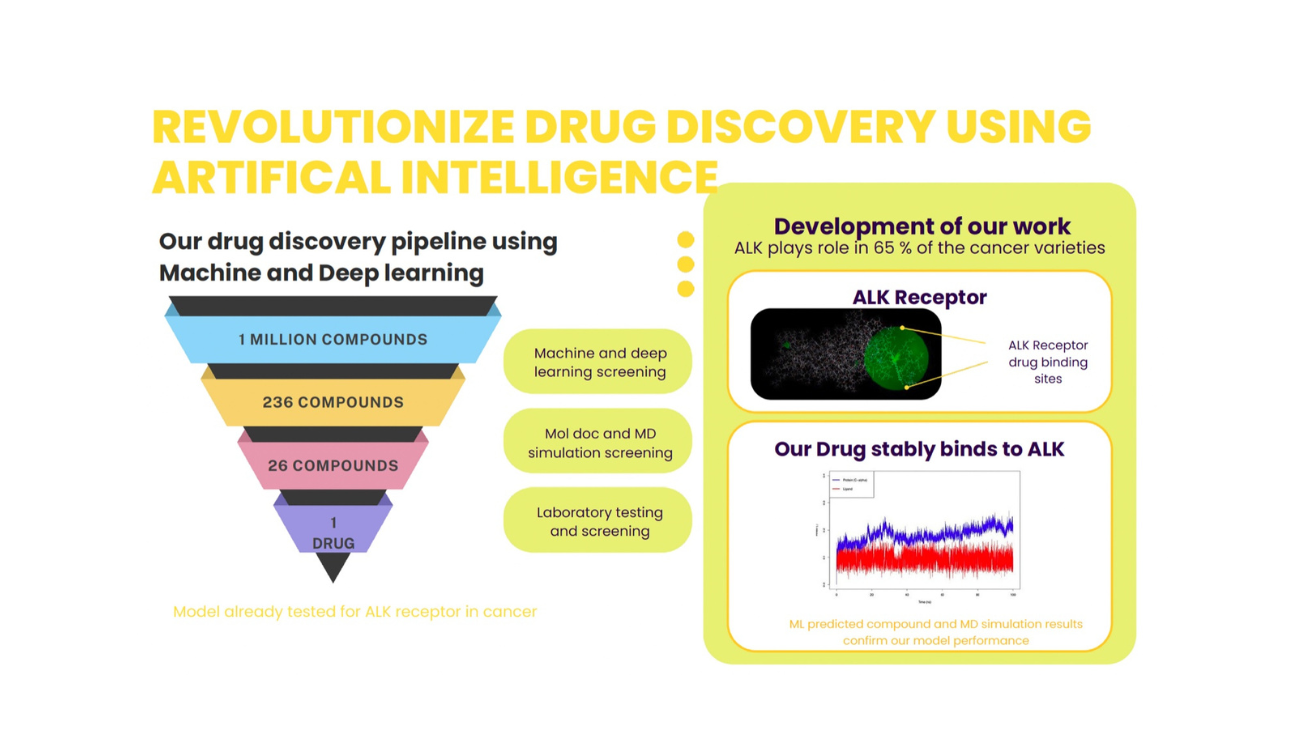
Machine Learning in Action: Our Approach
At the core of our project lies a sophisticated ML-powered drug discovery pipeline designed to predict biological activity, optimize molecular properties, and simulate protein-ligand interactions.
Key Components of Our ML Drug Discovery Pipeline
Molecular Representation We convert chemical structures into SMILES strings and molecular graphs, enabling our models to digitally process and analyze thousands of compounds efficiently.
Deep Learning for Activity Prediction
Using advanced models like Graph Neural Networks (GNNs) and Recurrent Neural Networks (RNNs), we predict binding affinities, toxicity profiles, and ADMET properties of candidate molecules.
Virtual Screening & Docking
ML accelerates high-throughput screening, narrowing down top candidates for structure-based docking against validated biological targets.
Target Identification
By integrating genomic, proteomic, and gene expression data, we identify novel and high-impact therapeutic targets, especially those involved in complex diseases and resistance pathways.
LigPlot Interaction Diagrams
We use LigPlot+ to visualize detailed protein-ligand interactions post-docking. This offers a 2D schematic view of hydrogen bonds and hydrophobic contacts, allowing researchers to interpret binding modes clearly and refine lead compounds.
Molecular Dynamics (MD) Simulations
To ensure stability and validate the biological relevance of docked complexes, we perform MD simulations. These simulations help assess the conformational flexibility, binding energy, and interaction dynamics of protein-ligand complexes over time in realistic environments. Join Us in Revolutionising Drug Discovery
At SMCS-Psi, we believe the future of medicine is data-driven, fast-tracked, and intelligent. If you’re a researcher, biotech entrepreneur, or pharma innovator looking to accelerate your R&D, we welcome collaborations and partnerships.
Contact us today to learn more about our machine learning and simulation solutions for drug discovery—or to explore how we can support your next breakthrough.
SMCS-Psi Data Analytics Pvt. Ltd.
Where Biology Meets Data Science.
Ravet, Pune (India) | Leipzig (Germany)
info@smcs-psi.com
www.smcs-psi.com
